Project 3. Modeling malignant myelopoiesis to increase efficacy of targeted leukemia therapy
PI: Richard Van Etten; Key Personnel: Kai Kessenbrock, Natalia Komarova, John S. Lowengrub, Qing Nie, Dominik Wodarz, Xiaohui Xie, Nilamani Jena
Chronic myeloid leukemia (CML), one of the most prevalent of human leukemias, is a natural model of dysregulated granulopoiesis driven by a single genetic abnormality in a hematopoietic stem cell, the BCR-ABL1 gene fusion. Although therapy with tyrosine kinase inhibitors (TKIs) such as imatinib mesylate has dramatically lowered the death rate in CML, lifelong treatment is needed and the associated economic costs are significant. Two major unaddressed questions are to understand the mechanism of primary resistance to TKI therapy (affecting ~10-15% of newly diagnosed CML patients), and to identify strategies to increase the frequency of complete molecular remission (CMR) in patients treated with TKIs and subsequently the rate of treatment-free remission (TFR), which may represent a surrogate for permanent cure of the disease. The scientific premise of this project is that new and clinically relevant insights into the biology of CML and its response to therapy can be gained by a more physiologically accurate mathematical model of the disease.
While mathematical models of CML have been developed by several groups, these models tend to be highly simplified and largely omit feedback interactions among the different hematopoietic components. These models are designed to fit clinical data sets of the response of patient populations to TKI therapy but unfortunately, they have not proven useful for understanding primary TKI resistance or for predicting TFR. In preliminary work, we found that nonlinear models that incorporate feedback are more robust, have a better fit to alternative patterns of patient response than simple linear models used previously, and allow predictions about the effects of interventions affecting parameters (such as cell cycle status) subject to feedback mechanisms on the response to TKI therapy. Preliminary analyses from prototype feedback models have already raised two provocative hypotheses about CML. The first is that the initial response to TKI therapy may depend on the relative size of the leukemic stem cell clone, which will be analyzed by TKI treatment of mice engrafted with different levels of BCR-ABL1+ stem cells. The second is that interventions that increase leukemic stem cell cycling may sensitize this population to killing by TKIs.
In this project, Van Etten and his team seek to develop improved mathematical models of chronic phase CML and the response to TKI therapy, to validate these models using data from a binary transgenic mouse model of CML and from human CML patients, and to utilize the models to test several hypotheses about the response of CML to therapy and to predict strategies for improving the treatment-free remission rate in CML. We address these issues by 1) Developing data-driven, dynamic models of CML hematopoiesis incorporating feedback control; 2) Measuring granulocytopoiesis parameters in a CML mouse model and in human CML patients; 3) Testing model-driven hypotheses about the response of CML to therapy and informing strategies for improving the treatment-free remission rate in CML.
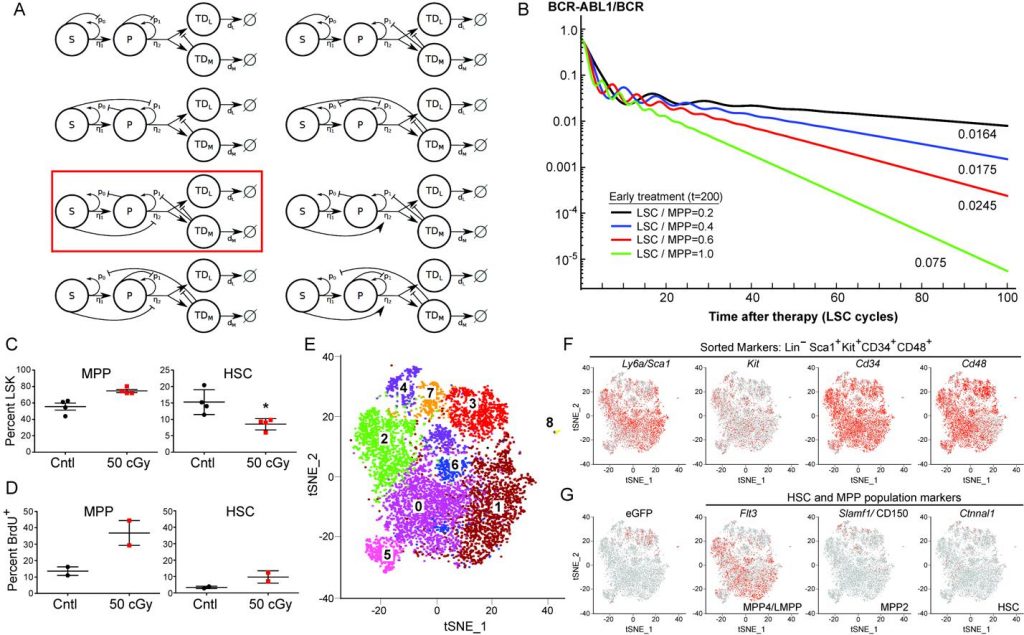
Figure 3. (A) Illustrative nonlinear feedback models of hematopoiesis. The red box indicates the model used in (B). (B) Increased leukemic stem cell (LSC) cycling predicts faster LSC decline on TKI therapy. The results correspond to different LSC cycling parameters applied (expressed as ratio of LSC to multipotential progenitor (MPP) cycling rate). Note the increasing negative slope of the second linear phase with increased cycling. (C,D) Selective decrement of HSC compartment increases MPP proliferation. Mice were irradiated (50 cGy), injected 24h post-radiation with BrdU and analyzed 12h later. (C) 50 cGy irradiation significantly (P=0.018) decreased the size (%LSK) of the HSC compartment (right panel) at 36h without a significant effect on the MPP compartment (left panel). (D) Reduction in HSC pool by low-dose radiation dramatically increases proliferation of the MPP compartment, supporting the existence of a negative feedback loop in the model above. (E) t-SNE analysis of scRNA-seq data from the MPP population isolated from mice with BCR-ABL1-induced CML shows eight separate subpopulations. (F) Expression of sorted lineage markers mapped on the t-SNE plot from (E). (G) Expression of GFP (marking leukemic MPP) and MPP/HSC markers mapped onto the t-SNE plot from (E).
Machine-based automated model selection methods are being utilized to arrive at a mathematical model that maintains appropriate stability and homeostasis, responds physiologically to stress and depletion of different cell compartments, and conforms to the limited existing qualitative data on CML hematopoiesis derived from mouse models and patient studies. To validate and inform potential models, binary/conditional BCR-ABL1 transgenic donor mice are being used to generate mixed BM chimeras via transplantation of high doses of unfractionated marrow cells without use of conditioning radiation. Recipients bearing a clone of BCR-ABL1+ cells have leukemia induced by withdrawal of doxycycline, leukemic mice are treated with BrdU or subjected to short-term stable isotope labeling with D2-glucose, marrow and spleen stem and progenitor compartments isolated by flow cytometry, and cell cycle and kinetic parameters estimated to inform the mathematical models. Single cell transcriptome profiling is used to investigate the heterogeneity of the MPP compartment and to discover potential regulatory mechanisms mediated by cytokine/receptor interactions. Feedback relationships are tested via direct in vivo manipulation using depleting monoclonal antibodies and through a novel mouse that targets metronidazole cytotoxicity to specific cell compartments. Parallel studies of human CML progenitor flux will be carried out through a clinical protocol of short-term D2-glucose labeling in patients presenting with suspected CML prior to diagnostic BM biopsy. The response to TKI therapy is also being analyzed. The integration of this work with Projects 1 and 2 occurs through the use of scRNA-seq as a tool for hypothesis generation and development and through the application of nonlinear mathematical models that have similar underlying structures (cell state transitions, proliferation and quiescence, positive and negative feedback), although the models in Project 3 are non-spatial.